
Our Health Library information does not replace the advice of a doctor. Please be advised that this information is made available to assist our patients to learn more about their health. Our providers may not see and/or treat all topics found herein.
Topic Contents
- General Information About Adult Central Nervous System (CNS) Tumors
- World Health Organization (WHO) Classification of Adult Primary CNS Tumors
- Treatment Option Overview for Adult Primary CNS Tumors
- Treatment of Primary CNS Tumors by Tumor Type
- Treatment of Primary Tumors of the Spinal Axis
- Metastatic Brain Tumors
- Treatment of Recurrent Adult CNS Tumors
- Latest Updates to This Summary (03 / 06 / 2024)
- About This PDQ Summary
Adult Central Nervous System Tumors Treatment (PDQ®): Treatment - Health Professional Information [NCI]
This information is produced and provided by the National Cancer Institute (NCI). The information in this topic may have changed since it was written. For the most current information, contact the National Cancer Institute via the Internet web site at http://cancer.gov or call 1-800-4-CANCER.
General Information About Adult Central Nervous System (CNS) Tumors
Incidence and Mortality
Brain tumors account for 85% to 90% of all primary central nervous system (CNS) tumors.[1] Estimated new cases and deaths from brain tumors and other nervous system tumors in the United States in 2024:[2]
- New cases: 25,400.
- Deaths: 18,760.
Data from the Surveillance, Epidemiology, and End Results (SEER) Program database for 2016 to 2020 indicated that the combined incidence of brain and other CNS tumors in the United States was 6.2 per 100,000 people per year, and the mortality rate was 4.4 deaths per 100,000 people per year.[3] Worldwide, approximately 308,102 new cases of brain and other CNS tumors were diagnosed in the year 2020, with an estimated 251,329 deaths.[4]
In general, the incidence of primary CNS tumors is higher in White individuals than in Black individuals, and mortality is higher in men than in women.[3]
Primary brain tumors include the following in decreasing order of frequency:[1]
- Anaplastic astrocytomas and glioblastomas (38% of primary brain tumors).
- Meningiomas and other mesenchymal tumors (27% of primary brain tumors).
- Pituitary tumors.
- Schwannomas.
- CNS lymphomas.
- Oligodendrogliomas.
- Ependymomas.
- Low-grade astrocytomas.
- Medulloblastomas.
Primary spinal tumors include the following in decreasing order of frequency:
- Schwannomas, meningiomas, and ependymomas (79% of primary spinal tumors).
- Sarcomas.
- Astrocytomas.
- Vascular tumors.
- Chordomas.
Primary brain tumors rarely spread to other areas of the body, but they can spread to other parts of the brain and to the spinal axis.
Anatomy
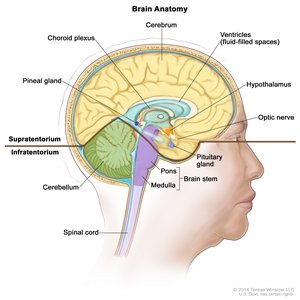
Anatomy of the inside of the brain. The supratentorium contains the cerebrum, ventricles (with cerebrospinal fluid shown in blue), choroid plexus, hypothalamus, pineal gland, pituitary gland, and optic nerve. The infratentorium contains the cerebellum and brain stem.
Risk Factors
Few definitive observations have been made about environmental or occupational causes of primary CNS tumors.[1]
The following potential risk factors have been considered:
- Exposure to vinyl chloride may be a risk factor for glioma.
- Epstein-Barr virus infection has been implicated in the etiology of primary CNS lymphoma.
- Transplant recipients and patients with AIDS have a substantially increased risk of primary CNS lymphoma.[1,5] For more information, see Primary CNS Lymphoma Treatment.
The familial tumor syndromes and related chromosomal abnormalities that are associated with CNS neoplasms include the following:[6,7]
- Neurofibromatosis type 1 (17q11).
- Neurofibromatosis type 2 (22q12).
- von Hippel-Lindau disease (3p25-26).
- Tuberous sclerosis (9q34, 16p13).
- Li-Fraumeni syndrome (17p13).
- Turcot syndrome type 1 (3p21, 7p22).
- Turcot syndrome type 2 (5q21).
- Nevoid basal cell carcinoma syndrome (9q22.3).
Clinical Features
The clinical presentation of various brain tumors is best appreciated by considering the relationship of signs and symptoms to anatomy.[1]
General signs and symptoms include the following:
- Headaches.
- Seizures.
- Visual changes.
- Gastrointestinal symptoms such as loss of appetite, nausea, and vomiting.
- Changes in personality, mood, mental capacity, and concentration.
Seizures are a presenting symptom in approximately 20% of patients with supratentorial brain tumors and may antedate the clinical diagnosis by months to years in patients with slow-growing tumors. Among all patients with brain tumors, 70% with primary parenchymal tumors and 40% with metastatic brain tumors develop seizures at some time during the clinical course.[8]
Diagnostic Evaluation
All brain tumors, whether primary, metastatic, malignant, or benign, must be differentiated from other space-occupying lesions that can have similar clinical presentations, such as abscesses, arteriovenous malformations, and infarctions.[9]
Imaging tests
Contrast-enhanced computed tomography (CT) and magnetic resonance imaging (MRI) have complementary roles in the diagnosis of CNS neoplasms.[1,9,10]
- The speed of CT is desirable for evaluating clinically unstable patients. CT is superior for detecting calcifications, skull lesions, and hyperacute hemorrhages (bleeding less than 24 hours old) and helps direct differential diagnosis and immediate management.
- MRI has superior soft-tissue resolution. MRI can better detect isodense lesions, tumor enhancements, and associated findings such as edema, all phases of hemorrhagic states (except hyperacute), and infarctions. High-quality MRI is the diagnostic study of choice in the evaluation of intramedullary and extramedullary spinal cord lesions.[1]
In posttherapy imaging, single-photon emission computed tomography (SPECT) and positron emission tomography (PET) may be useful in differentiating tumor recurrence from radiation necrosis.[9]
Biopsy
Biopsy confirmation to corroborate the suspected diagnosis of a primary brain tumor is critical, whether before surgery by needle biopsy or at the time of surgical resection. The exception is cases in which the clinical and radiological evidence clearly points to a benign tumor, which could potentially be managed with active surveillance without biopsy or treatment. For other cases, radiological patterns may be misleading, and a definitive biopsy is needed to rule out other causes of space-occupying lesions, such as metastatic cancer or infection.
CT- or MRI-guided stereotactic techniques can be used to place a needle safely and accurately into almost all locations in the brain.
Prognostic Factors
Several genetic alterations have emerged as powerful prognostic factors in diffuse glioma (astrocytoma, oligodendroglioma, mixed glioma, and glioblastoma), and these alterations may guide patient management. Specific alterations include the following:
- DNA methylation of the MGMT gene promoter.
- Mutation of the IDH1 or IDH2 gene.
- Codeletion of chromosomes 1p and 19q.
Other prognostic factors that confer poor prognosis include the following:[11,12]
- Age older than 40 years.
- Progressive disease.
- Tumor size larger than 5 cm.
- Tumor crossing the midline.
- Contrast enhancement on MRI.
- World Health Organization performance status (≥1).
- Neurological symptoms.
- Less than a gross total resection.
In an exploratory analysis of 318 patients with low-grade glioma treated with either radiation therapy alone or temozolomide chemotherapy alone, a combination of these prognostic factors demonstrated the following:[11]
- Longer progression-free survival (PFS) in patients with an IDH mutation without codeletion of 1p/19q when treated with radiation therapy (hazard ratio, 1.86; 95% confidence interval, 1.21–2.87; log-rank, P = .0043).
- No significant treatment-dependent differences in PFS for patients with an IDH mutation with codeletion of 1p/19q and IDH wild-type tumors.
- Patients with wild-type IDH tumors had the worst prognosis independent of treatment type.
- Patients with IDH-mutated tumors with codeletion of 1p/19q had the best prognosis.
- The O6-methylguanine-DNA methyltransferase (MGMT) promoter status in low-grade tumors was methylated in:
- All IDH mutations with codeletion of 1p/19q (45/45).
- Most, but not all (86%, 62/72), of the IDH mutations without codeletion of 1p/19q.
- Fifty-six percent (5/9) of the IDH wild-type cases.
For more information, see the Treatment of Primary Central Nervous System Tumors by Tumor Type section.
References:
- Mehta M, Vogelbaum MA, Chang S, et al.: Neoplasms of the central nervous system. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Lippincott Williams & Wilkins, 2011, pp 1700-49.
- American Cancer Society: Cancer Facts and Figures 2024. American Cancer Society, 2024. Available online. Last accessed June 21, 2024.
- National Cancer Institute: SEER Cancer Stat Facts: Brain and Other Nervous System Cancer. Bethesda, Md: National Cancer Institute. Available online. Last accessed March 5, 2024.
- Sung H, Ferlay J, Siegel RL, et al.: Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 71 (3): 209-249, 2021.
- Schabet M: Epidemiology of primary CNS lymphoma. J Neurooncol 43 (3): 199-201, 1999.
- Behin A, Hoang-Xuan K, Carpentier AF, et al.: Primary brain tumours in adults. Lancet 361 (9354): 323-31, 2003.
- Kleihues P, Cavenee WK, eds.: Pathology and Genetics of Tumours of the Nervous System. International Agency for Research on Cancer, 2000.
- Cloughesy T, Selch MT, Liau L: Brain. In: Haskell CM: Cancer Treatment. 5th ed. WB Saunders Co, 2001, pp 1106-42.
- Hutter A, Schwetye KE, Bierhals AJ, et al.: Brain neoplasms: epidemiology, diagnosis, and prospects for cost-effective imaging. Neuroimaging Clin N Am 13 (2): 237-50, x-xi, 2003.
- Ricci PE: Imaging of adult brain tumors. Neuroimaging Clin N Am 9 (4): 651-69, 1999.
- Baumert BG, Hegi ME, van den Bent MJ, et al.: Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 17 (11): 1521-1532, 2016.
- Reijneveld JC, Taphoorn MJ, Coens C, et al.: Health-related quality of life in patients with high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 17 (11): 1533-1542, 2016.
World Health Organization (WHO) Classification of Adult Primary CNS Tumors
This classification is based on the World Health Organization (WHO) classification of central nervous system (CNS) tumors.[1] The WHO approach incorporates and interrelates morphology, cytogenetics, molecular genetics, and immunological markers in an attempt to construct a cellular classification that is universally applicable and prognostically valid. Earlier attempts to develop a TNM (tumor, node, metastasis)-based classification were dropped for the following reasons:[2]
- Tumor size (T) is less relevant than are tumor histology and location.
- Nodal status (N) does not apply because the brain and spinal cord have no lymphatics.
- Metastatic spread (M) rarely applies because most patients with CNS neoplasms do not live long enough to develop metastatic disease.
The WHO grading of CNS tumors establishes a malignancy scale based on histological features of the tumor.[3] The histological grades are as follows:
- WHO grade I includes lesions with low proliferative potential, a frequently discrete nature, and the possibility of cure following surgical resection alone.
- WHO grade II includes lesions that are generally infiltrating and low in mitotic activity but recur more frequently than do grade I malignant tumors after local therapy. Some tumor types tend to progress to higher grades of malignancy.
- WHO grade III includes lesions with histological evidence of malignancy, including nuclear atypia and increased mitotic activity. These lesions have anaplastic histology and infiltrative capacity. They are usually treated with aggressive adjuvant therapy.
- WHO grade IV includes lesions that are mitotically active, necrosis prone, and generally associated with a rapid preoperative and postoperative progression and fatal outcomes. The lesions are usually treated with aggressive adjuvant therapy.
Table 1 lists the tumor types and grades.[4] Tumors limited to the peripheral nervous system are not included. Histopathology, grading methods, incidence, and what is known about etiology specific to each tumor type have been described in detail elsewhere.[4,5]
| I | II | III | IV | |
|---|---|---|---|---|
| a Reprinted with permission from Louis, DN, Ohgaki H, Wiestler, OD, Cavenee, WK.World Health Organization Classification of Tumours of the Central Nervous System. IARC, Lyon, 2007. | ||||
| Astrocytic tumors | ||||
| Subependymal giant cell astrocytoma | X | |||
| Pilocytic astrocytoma | X | |||
| Pilomyxoid astrocytoma | X | |||
| Diffuse astrocytoma | X | |||
| Pleomorphic xanthoastrocytoma | X | |||
| Anaplastic astrocytoma | X | |||
| Glioblastoma | X | |||
| Giant cell glioblastoma | X | |||
| Gliosarcoma | X | |||
| Oligodendroglial tumors | ||||
| Oligodendroglioma | X | |||
| Anaplastic oligodendroglioma | X | |||
| Oligoastrocytic tumors | ||||
| Oligoastrocytoma | X | |||
| Anaplastic oligoastrocytoma | X | |||
| Ependymal tumors | ||||
| Subependymoma | X | |||
| Myxopapillary ependymoma | X | |||
| Ependymoma | X | |||
| Anaplastic ependymoma | X | |||
| Choroid plexus tumors | ||||
| Choroid plexus papilloma | X | |||
| Atypical choroid plexus papilloma | X | |||
| Choroid plexus carcinoma | X | |||
| Other neuroepithelial tumors | ||||
| Angiocentric glioma | X | |||
| Chordoid glioma of the third ventricle | X | |||
| Neuronal and mixed neuronal-glial tumors | ||||
| Gangliocytoma | X | |||
| Ganglioglioma | X | |||
| Anaplastic ganglioma | X | |||
| Desmoplastic infantile astrocytoma and ganglioglioma | X | |||
| Dysembryoplastic neuroepithelial tumor | X | |||
| Central neurocytoma | X | |||
| Extraventricular neurocytoma | X | |||
| Cerebellar liponeurocytoma | X | |||
| Paraganglioma of the spinal cord | X | |||
| Papillary glioneuronal tumor | X | |||
| Rosette-forming glioneural tumor of the fourth ventricle | X | |||
| Pineal tumors | ||||
| Pineocytoma | X | |||
| Pineal parenchymal tumor of intermediate differentiation | X | X | ||
| Pineoblastoma | X | |||
| Papillary tumor of the pineal region | X | X | ||
| Embryonal tumors | ||||
| Medulloblastoma | X | |||
| CNS primitive neuroectodermal tumor | X | |||
| Atypical teratoid/rhabdoid tumor | X | |||
| Tumors of the cranial and paraspinal nerves | ||||
| Schwannoma | X | |||
| Neurofibroma | X | |||
| Perineurioma | X | X | X | |
| Malignant peripheral nerve sheath tumor | X | X | X | |
| Meningeal tumors | ||||
| Meningioma | X | |||
| Atypical meningioma | X | |||
| Anaplastic/malignant meningioma | X | |||
| Hemangiopericytoma | X | |||
| Anaplastic hemangiopericytoma | X | |||
| Hemangioblastoma | X | |||
| Tumors of the sellar region | ||||
| Craniopharyngioma | X | |||
| Granular cell tumor of the neurohypophysis | X | |||
| Pituicytoma | X | |||
| Spindle cell oncocytoma of the adenohypophysis | X | |||
Genomic Alterations
Alterations in the BRAF, IDH1, and IDH2 genes, and genomic 1p/19q codeletion, appear to be hallmark aberrations in particular glioma subtypes. Assessment for the presence of these mutations aids diagnosis and prognosis and, with regard to 1p/19q codeletion, predicts for response to chemotherapy.
In pilocytic astrocytomas (WHO grade I), tandem duplication at 7q34 leading to a KIAA1549::BRAF fusion is found in approximately 70% of pilocytic astrocytomas.[6,7,8] An activating point mutation in BRAF (V600E) is found in an additional 5% to 9% of these tumors and in general, RAF alterations occur in approximately 80% of pilocytic astrocytomas.
BRAF V600E mutations are observed (in about 60%) of other benign glioma variants, including pleomorphic xanthoastrocytoma and ganglioglioma, while BRAF tandem duplications are not found in these variant glioma tumors.[9,10,11]
Most WHO grade II and III diffuse gliomas (astrocytomas, oligodendrogliomas, and oligoastrocytomas) and 5% to 10% of glioblastomas (WHO grade IV) harbor point mutations in the R132 position of IDH1 or, rarely, the analogous codon in IDH2 (R172).[12,13,14,15,16] The presence of an IDH1 or IDH2 mutation is a strong prognostic factor. Patients with these mutant tumors have significantly longer survival independent of WHO grade or histological subtype.
Deletion of chromosomes 1p and 19q occurs through a translocation event [17] and is common in oligodendrogliomas. 1p/19q codeletion is a powerful prognostic factor and may predict for response to chemotherapy. For more information, see the Anaplastic oligodendrogliomas treatment section.
These genetic alterations have potential diagnostic utility. Presence of the IDH1 and IDH2 mutations may distinguish diffuse gliomas from other glioma variants, which often have BRAF genetic alterations, and nonneoplastic reactive astrocytosis.[18] Most (90%) IDH mutations in gliomas result in an R132H substitution, which can be detected with a highly sensitive and specific monoclonal antibody. A rapid immunohistochemical analysis using the mutant-specific IDH1 antibody can aid diagnostic analysis.[19]
Other CNS tumors are associated with characteristic patterns of altered oncogenes, altered tumor suppressor genes, and chromosomal abnormalities. Familial tumor syndromes with defined chromosomal abnormalities are associated with gliomas.
References:
- Kleihues P, Cavenee WK, eds.: Pathology and Genetics of Tumours of the Nervous System. International Agency for Research on Cancer, 2000.
- Brain and Spinal Cord. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp. 857–69.
- Kleihues P, Burger PC, Scheithauer BW: The new WHO classification of brain tumours. Brain Pathol 3 (3): 255-68, 1993.
- Louis DN, Ohgaki H, Wiestler OD, et al.: The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114 (2): 97-109, 2007.
- Louis DN, Ohgaki H, Wiestler OD, et al., eds.: WHO Classification of Tumours of the Central Nervous System. 4th ed. IARC Press, 2007.
- Sievert AJ, Jackson EM, Gai X, et al.: Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain Pathol 19 (3): 449-58, 2009.
- Pfister S, Janzarik WG, Remke M, et al.: BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 118 (5): 1739-49, 2008.
- Jones DT, Kocialkowski S, Liu L, et al.: Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68 (21): 8673-7, 2008.
- Dias-Santagata D, Lam Q, Vernovsky K, et al.: BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One 6 (3): e17948, 2011.
- MacConaill LE, Campbell CD, Kehoe SM, et al.: Profiling critical cancer gene mutations in clinical tumor samples. PLoS One 4 (11): e7887, 2009.
- Parsons DW, Jones S, Zhang X, et al.: An integrated genomic analysis of human glioblastoma multiforme. Science 321 (5897): 1807-12, 2008.
- Yan H, Parsons DW, Jin G, et al.: IDH1 and IDH2 mutations in gliomas. N Engl J Med 360 (8): 765-73, 2009.
- Dubbink HJ, Taal W, van Marion R, et al.: IDH1 mutations in low-grade astrocytomas predict survival but not response to temozolomide. Neurology 73 (21): 1792-5, 2009.
- Sanson M, Marie Y, Paris S, et al.: Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27 (25): 4150-4, 2009.
- Hartmann C, Hentschel B, Wick W, et al.: Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol 120 (6): 707-18, 2010.
- Hartmann C, Meyer J, Balss J, et al.: Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118 (4): 469-74, 2009.
- Jenkins RB, Blair H, Ballman KV, et al.: A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 66 (20): 9852-61, 2006.
- Camelo-Piragua S, Jansen M, Ganguly A, et al.: A sensitive and specific diagnostic panel to distinguish diffuse astrocytoma from astrocytosis: chromosome 7 gain with mutant isocitrate dehydrogenase 1 and p53. J Neuropathol Exp Neurol 70 (2): 110-5, 2011.
- Capper D, Weissert S, Balss J, et al.: Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol 20 (1): 245-54, 2010.
Treatment Option Overview for Adult Primary CNS Tumors
Primary CNS Tumors
This section discusses general treatment modalities for primary central nervous system (CNS) tumors. For a description of specific treatment options for each tumor type, see the Treatment of Primary Central Nervous System Tumors by Tumor Type section.
Radiation therapy and chemotherapy options vary according to histology and anatomical site of the CNS tumor. For glioblastoma, combined modality therapy with resection, radiation, and chemotherapy is standard. Anaplastic astrocytomas, anaplastic oligodendrogliomas, and anaplastic oligoastrocytomas represent only a small proportion of CNS gliomas; therefore, phase III randomized trials restricted to these tumor types are not generally practical. The natural histories of these tumors are variable, depending on histological and molecular factors; therefore, treatment guidelines are evolving. Therapy involving surgically implanted carmustine-impregnated polymer wafers combined with postoperative external-beam radiation therapy (EBRT) may play a role in the treatment of high-grade (grades III and IV) gliomas in some patients.[1]
Treatment options for primary CNS tumors include the following:
- Surgery.
- Radiation therapy.
- Chemotherapy.
- Active surveillance.
- Supportive therapy.
Surgery
For most types of CNS tumors in most locations, complete or near-complete surgical removal is generally attempted, within the constraints of preserving neurological function and the patient's underlying health. This practice is based on observational evidence that survival is better in patients who undergo tumor resection than in those who have closed biopsy alone.[2,3] The benefit of resection has not been tested in randomized trials. Selection bias can enter into observational studies despite attempts to adjust for patient differences that guide the decision to resect the tumor; therefore, the actual difference in outcome between radical surgery and biopsy alone may not be as large as noted in the retrospective studies.[3]
An exception to the use of resection is the case of deep-seated tumors such as pontine gliomas, which are diagnosed on clinical evidence and treated without initial surgery approximately 50% of the time. In most cases, however, diagnosis by biopsy is preferred. Stereotactic biopsy can be used for lesions that are difficult to reach and resect.
The primary goals of surgical resection include the following:[4]
- To establish a histological diagnosis.
- To reduce intracranial pressure by removing as much tumor as is safely possible to preserve neurological function.
Total elimination of primary malignant intraparenchymal tumors by surgery alone is rarely achievable. Therefore, intraoperative techniques have been developed to reach a balance between removing as much tumor as is practical and preserving functional status. For example, craniotomies with stereotactic resections of primary gliomas can be performed in cooperative patients while they are awake, with real-time assessment of neurological function.[5] Examples of intraoperative neurological assessment include the following:
- Resection proceeds until either the magnetic resonance imaging (MRI) signal abnormality being used to monitor the extent of surgery is completely removed or subtle neurological dysfunction appears (e.g., a slight decrease in rapid alternating motor movement or anomia).
- When the tumor is located in or near language centers in the cortex, intraoperative language mapping can be performed by electrode discharge-induced speech arrest while the patient is asked to count or read.[6]
As is the case with several other specialized operations [7,8] in which postoperative mortality has been associated with the number of procedures performed, postoperative mortality after surgery for primary brain tumors may be associated with hospital and/or surgeon volume.[9] Using the Nationwide Inpatient Sample hospital discharge database for the years 1988 to 2000, which represented 20% of inpatient admissions to nonfederal U.S. hospitals, investigators observed the following:[9]
- Large-volume hospitals had lower in-hospital mortality rates after craniotomies for primary brain tumors (odds ratio [OR], 0.75 for a tenfold higher caseload; 95% confidence interval [CI], 0.62–0.90) and after needle biopsies (OR, 0.54; 95% CI, 0.35–0.83).
- Although there was no specific sharp threshold in all-cause mortality outcomes between low-volume hospitals and high-volume hospitals, craniotomy-associated in-hospital mortality was 4.5% for hospitals with 5 or fewer procedures per year and 1.5% for hospitals with at least 42 procedures per year.
- In-hospital mortality rates decreased over the study years (perhaps because the proportion of elective nonemergent operations increased from 45% to 57%), but the decrease was more rapid in high-volume hospitals than in low-volume hospitals.
- High-volume surgeons had lower in-hospital patient mortality rates after craniotomy (OR, 0.60; 95% CI, 0.45–0.79).
As with any study of volume-outcome associations, these results may not be causal because of residual confounding factors such as referral patterns, private insurance, and patient selection, despite multivariable adjustment.
Radiation therapy
High-grade tumors
Radiation therapy has a major role in the treatment of patients with high-grade gliomas.
Evidence (postoperative radiation therapy [PORT]):
- A systematic review and meta-analysis of five randomized trials (plus one trial with allocation by birth date) comparing PORT with no radiation therapy showed a statistically significant survival advantage with radiation (risk ratio, 0.81; 95% CI, 0.74–0.88).[10][Level of evidence A1]
- A randomized trial comparing 60 Gy (in 30 fractions over 6 weeks) with 45 Gy (in 25 fractions over 4 weeks) showed superior survival in the first group (12 months vs. 9 months median survival; hazard ratio [HR], 0.81; 95% CI, 0.66–0.99). The accepted standard dose of EBRT for malignant gliomas is 60 Gy.[11][Level of evidence A1]
EBRT using either 3-dimensional conformal radiation therapy (3D-CRT) or intensity-modulated radiation therapy (IMRT) is considered an acceptable technique in radiation therapy delivery. Typically used are 2- to 3-cm margins on the MRI-based volumes (T1-weighted and fluid-attenuated inversion recovery [FLAIR]) to create the planning target volume.
Dose escalation using radiosurgery has not improved outcomes. A randomized trial tested radiosurgery as a boost added to standard EBRT, but the trial found no improvement in survival, quality of life, or patterns of relapse compared with EBRT without the boost.[12,13]
Brachytherapy has been used to deliver high doses of radiation locally to the tumor while sparing normal brain tissue. However, this approach is technically demanding and is less common since the advent of 3D-CRT and IMRT.
Low-grade tumors
Treatment options for patients with low-grade gliomas (i.e., low-grade astrocytomas, oligodendrogliomas, and mixed oligoastrocytomas) are not as clear as in the case of high-grade tumors and include observation, PORT, and chemotherapy with temozolomide.
Evidence (PORT vs. observation):
- The European Organisation for Research and Treatment of Cancer (EORTC) randomly assigned 311 patients with low-grade gliomas to undergo either radiation or observation in the EORTC-22845 trial.[14,15] On review of central pathology, about 25% of patients in the trial were reported to have high-grade tumors. Most of the control patients received radiation therapy at the time of progression.
- After a median follow-up of 93 months, the median progression-free survival (PFS) was 5.3 years in the radiation arm versus 3.4 years in the control arm (HR, 0.59; 95% CI, 0.45–0.77).[14,15][Level of evidence B1]
- There was no difference in the overall survival (OS). The median survival was 7.4 years in the radiation arm and 7.2 years in the control arm (HR, 0.97; 95% CI, 0.71–1.34; P = .87).[14,15][Level of evidence A1] This was caused by a longer survival after progression in the control arm (3.4 years) than in the radiation arm (1.0 year) (P < .0001).
- The investigators did not collect reliable quality-of-life measurements, so it is not clear whether the delay in initial relapse in the radiation therapy arm translated into improved function or quality of life.
Evidence (PORT versus temozolomide for patients with low-grade World Health Organization [WHO] grade II tumors with at least one high-risk feature):
- The EORTC 22033-26033 trial (NCT00182819) included 707 patients with low-grade glioma (WHO grade II astrocytoma, oligoastrocytoma, or oligodendroglioma) and at least one high-risk feature (age >40 years, progressive disease, tumor size >5 cm, tumor crossing the midline, or neurological symptoms). Patients were randomly assigned to receive either radiation therapy (n = 240) or temozolomide chemotherapy (n = 237). Radiation therapy consisted of conformal treatment (up to 50.4 Gy; 28 doses of 1.8 Gy daily, 5 days a week, for up to 6.5 weeks). Chemotherapy was dose-dense oral temozolomide (75 mg/m2 daily for 21 days, repeated every 28 days [one cycle], for a maximum of 12 cycles).[16,17]
- There was no significant difference in PFS (primary end point) or health-related quality of life (secondary end point).
- At a median follow-up of 48 months (interquartile range, 31–56), median PFS was 39 months (95% CI, 35–44) in the temozolomide group and 46 months (95% CI, 40–56) in the radiation therapy group (unadjusted HR, 1.16; 95% CI, 0.9–1.5; P = .22).[16][Level of evidence B1]
- An exploratory analysis of 318 molecularly defined patients found that patients with an IDH gene mutation without codeletion of 1p/19q displayed a significantly longer PFS when treated with radiation therapy (HR, 1.86; 95% CI, 1.21–2.87; log-rank P = .0043).
- There were no significant treatment-dependent differences in PFS for patients with IDH mutation with codeletion of 1p/19q and IDH wild-type tumors.
- Patients with wild-type IDH tumors had the worst prognosis independent of treatment type.
- Patients with IDH-mutated tumors with codeletion of 1p/19q had the best prognosis.
- The O6-methylguanine-DNA methyltransferase (MGMT) promoter status was methylated in the following:
- All IDH mutations with codeletion of 1p/19q (45/45).
- Sixty-two of 72 (86%) of the IDH mutations without codeletion of 1p/19q.
- Five of nine (56%) of the IDH wild-type cases.
Disease progression, subsequent neoplasms, or recurrences
There are no randomized trials to delineate the role of repeat radiation after disease progression or the development of radiation-induced cancers. The literature is limited to small retrospective case series, which makes interpretation difficult.[18] The decision to repeat radiation must be made carefully because of the risk of neurocognitive deficits and radiation-induced necrosis. One advantage of radiosurgery is the ability to deliver therapeutic doses to recurrent tumors that may require the re-irradiation of previously irradiated brain tissue beyond tolerable dose limits.
Chemotherapy
Systemic chemotherapy
For many years, the nitrosourea carmustine ([bis-chloroethylnitrosourea] BCNU) was the standard chemotherapy agent added to surgery and radiation therapy for malignant gliomas, based on the Radiation Therapy Oncology Group's (RTOG's) randomized trial (RTOG-8302).[19][Level of evidence A1] A modest impact on survival with the use of nitrosourea-containing chemotherapy regimens for malignant gliomas was confirmed in a patient-level meta-analysis of 12 randomized trials (combined HRdeath, 0.85; 95% CI, 0.78–0.91).[20]
A large multicenter trial (NCT00006353) of patients with glioblastoma, conducted by the EORTC-National Cancer Institute of Canada, reported a survival advantage with the use of temozolomide in addition to radiation therapy.[21,22][Level of evidence A1] On the basis of these results, the oral agent temozolomide has replaced BCNU as the standard systemic chemotherapy for malignant gliomas. For more information, see the Glioblastomas treatment section.
Long-term results of randomized trials in high-risk, low-grade (WHO grade II) gliomas [23][Level of evidence A1] and anaplastic (WHO grade III) oligodendroglial tumors [24,25][Level of evidence A1] have demonstrated that the addition of procarbazine, lomustine, and vincristine (PCV) chemotherapy to radiation therapy after surgery extends survival. Radiation and PCV chemotherapy should be considered for patients deemed appropriate for therapy. For more information, see the Treatment of Primary Central Nervous System Tumors by Tumor Type section.
Localized chemotherapy (carmustine wafer)
The ability to give high doses of chemotherapy while avoiding systemic toxicity is desirable because malignant glioma–related deaths are usually due to uncontrolled intracranial disease rather than distant metastases. A biodegradable carmustine wafer has been developed for that purpose. The wafers contain 3.85% carmustine, and up to eight wafers are implanted into the tumor bed lining at the time of open resection, with an intended total dose of about 7.7 mg per wafer (61.6 mg maximum per patient) over a period of 2 to 3 weeks.
Two randomized placebo-controlled trials of this focal drug-delivery method have shown an OS advantage associated with the carmustine wafers versus radiation therapy alone. In both trials, the upper age limit for patients was 65 years.
Evidence (carmustine wafer):
- A small trial was closed because of a lack of continued availability of the carmustine wafers after 32 patients with high-grade gliomas had been entered.[26]
- Although OS was better in the carmustine-wafer group (median 58.1 vs. 39.9 weeks; P = .012), there was an imbalance in the study arms (only 11 of 16 patients in the carmustine-wafer group vs. 16 of the 16 patients in the placebo-wafer group had grade IV glioblastoma tumors).
- A multicenter study of 240 patients with primary malignant gliomas, 207 of whom had glioblastoma, was more informative.[27,28] At initial surgery, patients received either carmustine wafers or placebo wafers, followed by radiation therapy (55–60 Gy). Systemic therapy was not allowed until recurrence, except in the case of anaplastic oligodendrogliomas (n = 9). Unlike the initial trial, patient characteristics were well balanced between the study arms.
- Median survival in the two groups was 13.8 months in patients treated with carmustine wafers versus 11.6 months in placebo-treated patients (HR, 0.73; 95% CI, 0.56–0.96; P = .017).
- A systematic review combining both studies [26,27,28] estimated an HR for overall mortality of 0.65; 95% CI, 0.48–0.86; P = .003.[29][Level of evidence A1]
Active surveillance
Active surveillance is appropriate in some circumstances. With the increasing use of sensitive neuroimaging tools, detection of asymptomatic low-grade meningiomas has increased; most appear to show minimal growth and can often be safely observed, with therapy deferred until the detection of tumor growth or the development of symptoms.[30,31]
Supportive therapy
Dexamethasone, mannitol, and furosemide are used to treat the peritumoral edema associated with brain tumors. The use of anticonvulsants is mandatory for patients with seizures.[4]
References:
- Lallana EC, Abrey LE: Update on the therapeutic approaches to brain tumors. Expert Rev Anticancer Ther 3 (5): 655-70, 2003.
- Laws ER, Parney IF, Huang W, et al.: Survival following surgery and prognostic factors for recently diagnosed malignant glioma: data from the Glioma Outcomes Project. J Neurosurg 99 (3): 467-73, 2003.
- Chang SM, Parney IF, Huang W, et al.: Patterns of care for adults with newly diagnosed malignant glioma. JAMA 293 (5): 557-64, 2005.
- Cloughesy T, Selch MT, Liau L: Brain. In: Haskell CM: Cancer Treatment. 5th ed. WB Saunders Co, 2001, pp 1106-42.
- Meyer FB, Bates LM, Goerss SJ, et al.: Awake craniotomy for aggressive resection of primary gliomas located in eloquent brain. Mayo Clin Proc 76 (7): 677-87, 2001.
- Sanai N, Mirzadeh Z, Berger MS: Functional outcome after language mapping for glioma resection. N Engl J Med 358 (1): 18-27, 2008.
- Begg CB, Cramer LD, Hoskins WJ, et al.: Impact of hospital volume on operative mortality for major cancer surgery. JAMA 280 (20): 1747-51, 1998.
- Birkmeyer JD, Finlayson EV, Birkmeyer CM: Volume standards for high-risk surgical procedures: potential benefits of the Leapfrog initiative. Surgery 130 (3): 415-22, 2001.
- Barker FG, Curry WT, Carter BS: Surgery for primary supratentorial brain tumors in the United States, 1988 to 2000: the effect of provider caseload and centralization of care. Neuro Oncol 7 (1): 49-63, 2005.
- Laperriere N, Zuraw L, Cairncross G, et al.: Radiotherapy for newly diagnosed malignant glioma in adults: a systematic review. Radiother Oncol 64 (3): 259-73, 2002.
- Bleehen NM, Stenning SP: A Medical Research Council trial of two radiotherapy doses in the treatment of grades 3 and 4 astrocytoma. The Medical Research Council Brain Tumour Working Party. Br J Cancer 64 (4): 769-74, 1991.
- Tsao MN, Mehta MP, Whelan TJ, et al.: The American Society for Therapeutic Radiology and Oncology (ASTRO) evidence-based review of the role of radiosurgery for malignant glioma. Int J Radiat Oncol Biol Phys 63 (1): 47-55, 2005.
- Souhami L, Seiferheld W, Brachman D, et al.: Randomized comparison of stereotactic radiosurgery followed by conventional radiotherapy with carmustine to conventional radiotherapy with carmustine for patients with glioblastoma multiforme: report of Radiation Therapy Oncology Group 93-05 protocol. Int J Radiat Oncol Biol Phys 60 (3): 853-60, 2004.
- Karim AB, Afra D, Cornu P, et al.: Randomized trial on the efficacy of radiotherapy for cerebral low-grade glioma in the adult: European Organization for Research and Treatment of Cancer Study 22845 with the Medical Research Council study BRO4: an interim analysis. Int J Radiat Oncol Biol Phys 52 (2): 316-24, 2002.
- van den Bent MJ, Afra D, de Witte O, et al.: Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet 366 (9490): 985-90, 2005.
- Baumert BG, Hegi ME, van den Bent MJ, et al.: Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 17 (11): 1521-1532, 2016.
- Reijneveld JC, Taphoorn MJ, Coens C, et al.: Health-related quality of life in patients with high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 17 (11): 1533-1542, 2016.
- Paulino AC, Mai WY, Chintagumpala M, et al.: Radiation-induced malignant gliomas: is there a role for reirradiation? Int J Radiat Oncol Biol Phys 71 (5): 1381-7, 2008.
- Walker MD, Green SB, Byar DP, et al.: Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N Engl J Med 303 (23): 1323-9, 1980.
- Stewart LA: Chemotherapy in adult high-grade glioma: a systematic review and meta-analysis of individual patient data from 12 randomised trials. Lancet 359 (9311): 1011-8, 2002.
- Stupp R, Mason WP, van den Bent MJ, et al.: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352 (10): 987-96, 2005.
- Stupp R, Hegi ME, Mason WP, et al.: Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10 (5): 459-66, 2009.
- Buckner JC, Pugh SL, Shaw EG, et al.: Phase III study of radiation therapy with or without procarbazine, CCNU, and vincristine (PCV) in low-grade glioma: RTOG 9802 with Alliance, ECOG, and SWOG. [Abstract] J Clin Oncol 32 (Suppl 5): A-2000, 2014.
- van den Bent MJ, Brandes AA, Taphoorn MJ, et al.: Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 31 (3): 344-50, 2013.
- Cairncross G, Wang M, Shaw E, et al.: Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol 31 (3): 337-43, 2013.
- Valtonen S, Timonen U, Toivanen P, et al.: Interstitial chemotherapy with carmustine-loaded polymers for high-grade gliomas: a randomized double-blind study. Neurosurgery 41 (1): 44-8; discussion 48-9, 1997.
- Westphal M, Hilt DC, Bortey E, et al.: A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro-oncol 5 (2): 79-88, 2003.
- Westphal M, Ram Z, Riddle V, et al.: Gliadel wafer in initial surgery for malignant glioma: long-term follow-up of a multicenter controlled trial. Acta Neurochir (Wien) 148 (3): 269-75; discussion 275, 2006.
- Hart MG, Grant R, Garside R, et al.: Chemotherapeutic wafers for high grade glioma. Cochrane Database Syst Rev (3): CD007294, 2008.
- Nakamura M, Roser F, Michel J, et al.: The natural history of incidental meningiomas. Neurosurgery 53 (1): 62-70; discussion 70-1, 2003.
- Yano S, Kuratsu J; Kumamoto Brain Tumor Research Group: Indications for surgery in patients with asymptomatic meningiomas based on an extensive experience. J Neurosurg 105 (4): 538-43, 2006.
Treatment of Primary CNS Tumors by Tumor Type
| Tumor Type | Treatment Options |
|---|---|
| Astrocytic tumors | |
| —Brain stem gliomas | Radiation therapy |
| —Pineal astrocytic tumors | Surgery plus radiation therapy |
| Surgery plus radiation therapy and chemotherapy for higher-grade tumors | |
| —Pilocytic astrocytomas | Surgery alone |
| Surgery followed by radiation therapy | |
| —Diffuse astrocytomas (WHO grade II) | Surgery with or without radiation therapy |
| Surgery followed by radiation therapy and chemotherapy | |
| —Anaplastic astrocytomas (WHO grade III) | Surgery plus radiation therapy with or without chemotherapy |
| Surgery plus chemotherapy | |
| —Glioblastomas | Surgery plus radiation therapy and chemotherapy |
| Surgery plus radiation therapy | |
| Carmustine-impregnated polymer implant | |
| Radiation therapy and concurrent chemotherapy | |
| Oligodendroglial tumors | |
| —Oligodendrogliomas | Surgery with or without radiation therapy |
| Surgery with radiation therapy and chemotherapy | |
| —Anaplastic oligodendrogliomas | Surgery plus radiation therapy with or without chemotherapy |
| Mixed gliomas | Surgery plus radiation therapy with or without chemotherapy |
| Ependymal tumors | |
| —Grades I and II ependymal tumors | Surgery alone |
| Surgery followed by radiation therapy | |
| —Anaplastic ependymoma | Surgery plus radiation therapy |
| Embryonal cell tumors | |
| —Medulloblastomas | Surgery plus craniospinal radiation therapy |
| Pineal parenchymal tumors | Surgery plus radiation therapy(for pineocytoma) |
| Surgery plus radiation therapy and chemotherapy(for pineoblastoma) | |
| Meningeal tumors | |
| —Grade I meningiomas | Active surveillance with deferred treatment |
| Surgery | |
| Stereotactic radiosurgery | |
| Surgery plus radiation therapy | |
| Fractionated radiation therapy | |
| —Grades II and III meningiomas and hemangiopericytomas | Surgery plus radiation therapy |
| Germ cell tumors | Depends on multiple factors |
| Tumors of the sellar region | |
| —Craniopharyngiomas | Surgery alone |
| Debulking surgery plus radiation therapy | |
Astrocytic Tumors Treatment
Brain stem gliomas treatment
Patients with brain stem gliomas have relatively poor prognoses that correlate with histology (when biopsies are performed), location, and extent of tumor. The overall median survival time of patients in studies has been 44 to 74 weeks.
Treatment options for brain stem gliomas include the following:
- Radiation therapy.
Pineal astrocytic tumors treatment
Depending on the degree of anaplasia, patients with pineal astrocytomas have variable prognoses. Patients with higher-grade tumors have worse prognoses.
Treatment options for pineal astrocytic tumors include the following:
- Surgery plus radiation therapy for pineal astrocytoma.
- Surgery plus radiation therapy and chemotherapy for higher-grade tumors.
Pilocytic astrocytomas treatment
This astrocytic tumor is classified as a World Health Organization (WHO) grade I tumor and is often curable.
Treatment options for pilocytic astrocytomas include the following:
- Surgery alone if the tumor is totally resectable.
- Surgery followed by radiation therapy to known or suspected residual tumor.
Diffuse astrocytomas treatment
This WHO grade II astrocytic tumor is less often curable than is a pilocytic astrocytoma.
Treatment options for diffuse astrocytomas (WHO grade II) include the following:
- Surgery with or without radiation therapy.
- Surgery followed by radiation therapy and chemotherapy.
Controversy exists about the timing of radiation therapy after surgery. For more information, see the Low-grade tumors section.
- Radiation therapy improved progression-free survival (PFS) in patients who received early radiation therapy in the European Organisation for Research and Treatment of Cancer (EORTC) EORTC-22845 trial. For more information, see the Oligodendrogliomas treatment section.[1][Level of evidence A1]
- In the same trial, there was no difference in overall survival (OS) between patients who had radiation therapy after surgery and those who were treated with radiation therapy at the time of progression.[1][Level of evidence A1]
Some physicians use surgery alone if a patient has clinical factors that are considered low risk, such as age younger than 40 years and the lack of contrast enhancement on a computed tomography scan.[2]
Evidence (surgery followed by radiation therapy and chemotherapy):
- For patients with low-grade (WHO grade II) tumors, which are considered high risk, radiation therapy followed by six cycles of vincristine (PCV) chemotherapy is a recommended option. This recommendation is based on the long-term follow-up results of the Radiation Therapy Oncology Group's (RTOG's) 1986-initiated randomized trial (RTOG 9802 [NCT00003375]).[3][Level of evidence A1] In this trial, patients with high-risk, low-grade glioma, defined as patients aged 18 to 39 years with biopsy or subtotal resection, or patients aged 40 years or older, were randomly assigned to either 54 Gy of radiation therapy or radiation therapy followed by six cycles of PCV chemotherapy.
- The addition of PCV to radiation therapy increased median PFS from 4.0 years to 10.4 years (hazard ratio [HR], 0.50; P = .002) and median OS from 7.8 years to 13.3 years (HR, 0.59; P = .03).
- Notably, the RTOG 9802 study enrolled patients with a variety of tumors, including astrocytomas, oligodendrogliomas, and mixed oligoastrocytomas.
- In a risk-adjusted multivariate analysis, patients treated with PCV and patients with an oligodendroglial histology had better survival outcomes. A subset analysis of histological type suggested that the addition of PCV mainly benefited patients with oligodendroglial tumors, although this data is yet to be validated.[4]
- Median OS for PCV versus the control arm was not reached versus 10.8 years for oligodendrogliomas (P = .008), 11.4 years versus 5.9 years for oligoastrocytomas (P = .05), and 7.7 years versus 4.4 years for astrocytomas (P = .31).
The discovery of the IDH1 and IDH2 mutations in diffuse gliomas has greatly helped to identify patients with high-risk disease. Large retrospective studies have demonstrated that IDH1 and IDH2 mutations are powerful independent prognostic factors for improved survival.[5,6,7,8,9] Most WHO grade II and III gliomas harbor the IDH1 and IDH2 mutations,[6,10,11] and, therefore, the presence of the IDH1 and IDH2 mutations should be included in the assessment of high risk. Molecular correlative data from the RTOG 98-02 trial, which would be informative about which patients benefited the most from the addition of PCV, have not been reported.
Anaplastic astrocytomas treatment
Patients with anaplastic astrocytomas (WHO grade III) have a low cure rate with standard local treatment.
Treatment options for anaplastic astrocytomas include the following:
- Surgery plus radiation therapy with or without chemotherapy.
- Surgery plus chemotherapy.
A subset of anaplastic astrocytomas is aggressive; these tumors are frequently managed in the same way as glioblastomas, with surgery and radiation, and often with chemotherapy. However, the optimal treatment for these tumors is not established. Two phase III randomized trials restricted to patients with anaplastic gliomas (NCT00626990 and NCT00887146) are active, but efficacy data are not available. It is not known whether the improved survival of patients with chemotherapy-treated glioblastoma can be extrapolated to patients with anaplastic astrocytomas.
The IDH1 and IDH2 mutations are present in 50% to 70% of anaplastic astrocytomas and are independently associated with significantly improved survival.[6,9] Assessment of the IDH1 and IDH2 mutation status may guide decisions about treatment options.
Evidence (surgery plus radiation therapy or chemotherapy):
- Postoperative radiation alone has been compared with postoperative chemotherapy alone in patients with anaplastic gliomas (i.e., 144 astrocytomas, 91 oligoastrocytomas, and 39 oligodendrogliomas), with crossover to the other modality at the time of tumor progression. Of the 139 patients randomly assigned to undergo radiation therapy, 135 were randomly assigned to receive chemotherapy, with a 32-week course of either PCV or single-agent temozolomide (2:1:1 randomization).[12][Levels of evidence A1 and B1]
- The order of the modalities did not affect time-to-treatment failure (TTF) or OS.
- Neither TTF nor OS differed across the treatment arms.
Patients with anaplastic astrocytomas are appropriate candidates for clinical trials designed to improve local control by adding newer forms of treatment to standard treatment. Information about ongoing clinical trials is available from the NCI website.
Glioblastomas treatment
For patients with glioblastoma (WHO grade IV), the cure rate is very low with standard local treatment.
Methylation of the promoter of the MGMT DNA repair enzyme gene is an independent prognostic factor for improved survival in newly diagnosed glioblastoma.[13,14]MGMT promoter methylation and concomitant inactivation of the DNA repair enzyme activities may also predict for response to temozolomide chemotherapy.[13] However, the clinical data that MGMT promoter methylation is a predictive marker is less certain.
Treatment options for patients with newly diagnosed glioblastoma include the following:
- Surgery plus radiation therapy and chemotherapy.
- Surgery plus radiation therapy.
- Carmustine-impregnated polymer implanted during initial surgery.
- Radiation therapy and concurrent chemotherapy.
The standard treatment for patients with newly diagnosed glioblastoma is surgery followed by concurrent radiation therapy and daily temozolomide, and then followed by six cycles of temozolomide. The addition of bevacizumab to radiation therapy and temozolomide did not improve OS.
Evidence (surgery plus radiation therapy and chemotherapy):
- Standard therapy is based on a large, multicenter, randomized trial (NCT00006353) conducted by the EORTC and National Cancer Institute of Canada (NCIC). This trial reported a survival benefit with concurrent radiation therapy and temozolomide, compared with radiation therapy alone.[15,16][Level of evidence A1] In this study, 573 patients with glioblastoma were randomly assigned to receive standard radiation to the tumor volume with a 2- to 3-cm margin (60 Gy, 2 Gy per fraction, over 6 weeks) alone or with temozolomide (75 mg/m2 orally per day during radiation therapy for up to 49 days, followed by a 4-week break and then up to six cycles of five daily doses every 28 days at a dose of 150 mg/m2, increasing to 200 mg/m2 after the first cycle).
- OS was statistically significantly better in the combined radiation therapy–temozolomide group (HRdeath, 0.6; 95% confidence interval [CI], 0.5–0.7; OS rate at 3 years was 16.0% for the radiation therapy–temozolomide group vs. 4.4% in the radiation therapy–alone group).
- A companion molecular correlation subset study to the EORTC-NCIC trial provided strong evidence that epigenetic silencing of the MGMT DNA-repair gene by promoter DNA methylation was associated with increased OS in patients with newly diagnosed glioblastoma.[13]
- MGMT promoter methylation was an independent favorable prognostic factor (HR, 0.45; 95% CI, 0.32–0.61; log-rank P < .001).
- The median OS for patients with MGMT methylation was 18.2 months (95% CI, 15.5–22.0), compared with 12.2 months (95% CI, 11.4–13.5) for patients without MGMT methylation.
- To test whether protracted (dose-dense) temozolomide enhances treatment response in patients with newly diagnosed glioblastoma, a multicenter, randomized, phase III trial conducted by the RTOG, EORTC, and the North Central Cancer Therapy Group, RTOG 0525 (NCT00304031), compared standard adjuvant temozolomide treatment (days 1–5 of a 28-day cycle) with a dose-dense schedule (days 1–21 of a 28-day cycle). All patients were treated with surgery followed by radiation therapy and concurrent daily temozolomide. Patients were then randomly assigned to receive either standard adjuvant temozolomide or dose-dense temozolomide.[14][Level of evidence A1]
- Among 833 randomly assigned patients, no statistically significant difference between standard and dose-dense temozolomide was observed for median OS (16.6 months for standard temozolomide vs. 14.9 months for dose-dense temozolomide; HR, 1.03; P = .63) or for median PFS (5.5 vs. 6.7 months; HR, 0.87; P = .06).
- Protracted temozolomide, which depletes intracellular MGMT, was predicted to have greater efficacy in tumors with MGMT-promoter methylation. To test this retrospectively, MGMT status was determined in 86% of randomly assigned patients. No difference in efficacy was observed in either the MGMT-methylated or MGMT-unmethylated subsets. There was no survival advantage for the use of dose-dense temozolomide versus standard-dose temozolomide in newly diagnosed glioblastoma patients, regardless of MGMT status. However, this study confirmed the strong prognostic effect of MGMT methylation because the median OS was 21.2 months (95% CI, 17.9–24.8) for patients with methylation versus 14 months (HR, 1.74; 95% CI, 12.9–14.7; P < .001) for patients without methylation.
- The efficacy of dose-dense temozolomide for patients who have recurrent glioblastoma, however, is yet to be determined.
Evidence (surgery and chemoradiation therapy with or without bevacizumab):
In 2013, final data from two multicenter, phase III, randomized, double-blind, placebo-controlled trials of bevacizumab in patients who had newly diagnosed glioblastoma were reported: RTOG 0825 (NCT00884741) and the Roche-sponsored AVAglio (NCT00943826).[17,18][Level of evidence A1] Bevacizumab did not improve OS in either trial.
There was significant crossover in both trials. Approximately 40% of RTOG 0825 patients and approximately 30% of AVAglio patients received bevacizumab at the first sign of disease progression.
- RTOG 0825 (NCT00884741): Patients were randomly assigned to receive standard therapy (chemoradiation therapy with temozolomide) or standard therapy plus bevacizumab. OS and PFS were coprimary end points.[17][Level of evidence A1]
- Bevacizumab did not improve OS (median OS was 16–17 months for each arm). However, it increased median PFS (10.7 months in the bevacizumab arm vs. 7.3 months in the placebo arm; HR, 0.79; P = .007).
- The PFS result in the RTOG 0825 trial did not meet the prespecified significance level (P = .004).
- AVAglio (NCT00943826): Patients were randomly assigned to receive standard therapy (chemoradiation therapy with temozolomide) or standard therapy plus bevacizumab. OS and PFS were coprimary end points.[18][Level of evidence A1]
- Bevacizumab did not improve OS (median OS was 16–17 months for each arm). However, it increased median PFS (10.6 months in the bevacizumab arm vs. 6.2 months in the placebo arm; HR, 0.64; P < .0001).
- The PFS result was statistically significant and associated with clinical benefit because patients who received bevacizumab remained functionally independent longer (9.0 months in the bevacizumab arm vs. 6.0 months in the standard therapy arm) and had a longer time until their Karnofsky Performance status deteriorated (HR, 0.65; P < .0001).
- Patients who received bevacizumab also had delayed initiation of corticosteroids (12.3 months vs. 3.7 months; HR, 0.71; P = .002), and more patients were able to discontinue corticosteroids if they were already taking them (66% in the bevacizumab arm vs. 47% in the standard therapy arm).
The two trials had contradictory results in health-related quality of life (HRQOL) and neurocognitive outcomes studies. In the mandatory HRQOL studies in the AVAglio trial, bevacizumab-treated patients experienced improved HRQOL, but bevacizumab-treated patients in the elective RTOG 0825 studies showed more decline in patient-reported HRQOL and neurocognitive function. The reasons for these discrepancies are unclear.
On the basis of these results, there is no definite evidence that the addition of bevacizumab to standard therapy is beneficial for all newly diagnosed glioblastoma patients. Certain subgroups may benefit from the addition of bevacizumab, but this is not yet known.
Patients with glioblastoma are appropriate candidates for clinical trials designed to improve local control by adding newer forms of treatment to standard treatment. Information about ongoing clinical trials is available from the NCI website.
Oligodendroglial Tumors Treatment
Oligodendrogliomas treatment
Patients who have oligodendrogliomas (WHO grade II) generally have better prognoses than do patients who have diffuse astrocytomas. In particular, patients who have oligodendrogliomas with 1p/19q codeletion have a much longer survival.[3] Most of the oligodendrogliomas eventually progress.
Treatment options for oligodendrogliomas include the following:
- Surgery with or without radiation therapy.
- Surgery with radiation therapy and chemotherapy.
Controversy exists concerning the timing of radiation therapy after surgery. A study (EORTC-22845) of 300 patients with low-grade gliomas who had surgery and were randomly assigned to either radiation therapy or watchful waiting, did not show a difference in OS between the two groups.[1][Level of evidence A1] For more information, see the Low-grade tumors section.
For low-grade (WHO grade II) tumors that are considered high risk, radiation therapy followed by six cycles of PCV chemotherapy is a recommended option based on the long-term follow-up results of RTOG-9802, a randomized trial for high-risk, low-grade gliomas.[3][Level of evidence A1] Notably, RTOG-9802 enrolled patients with a variety of tumors, including astrocytomas, oligodendrogliomas, and mixed oligoastrocytomas. In a retrospective subset analysis, only the oligodendroglial tumors appeared to benefit from the addition of PCV.[4]. For more information, see the Diffuse astrocytomas treatment section.
The discovery of the IDH1 and IDH2 mutations, which are independent prognostic factors for significantly improved survival in diffuse gliomas, has greatly helped to identify patients with high-risk disease. For more information, see the Diffuse astrocytomas treatment section. In addition, a high proportion of WHO grade II oligodendrogliomas have 1p/19q codeletion, which is a powerful prognostic factor for improved survival.[19,20,21] Therefore, the presence of the IDH1 and IDH2 mutations and 1p/19q codeletion should be included in the assessment of high risk. Molecular correlative data from the RTOG-9802 trial, which would be informative about which patients benefited most from the addition of PCV, have not been reported.
Anaplastic oligodendrogliomas treatment
Patients with anaplastic oligodendrogliomas (WHO grade III) have a low cure rate with standard local treatment, but their prognoses are generally better than are the prognoses of patients with anaplastic astrocytomas. Prognoses are particularly better for patients with 1p/19q codeletion, which occurs in most of these tumors. Two phase III randomized trials restricted to patients with anaplastic gliomas (NCT00626990 and NCT00887146) are active; however, efficacy data are not yet available. For more information, see the Anaplastic astrocytomas treatment section. These patients are appropriate candidates for clinical trials designed to improve local control by adding newer forms of treatment.
Information about ongoing clinical trials is available from the NCI website.
Treatment options for anaplastic oligodendrogliomas include the following:
- Surgery plus radiation therapy with or without chemotherapy.[22]
Evidence (surgery followed by radiation therapy with or without chemotherapy):
- Mature results from the EORTC Brain Tumor Group Study 26951 (NCT00002840), a phase III randomized study with 11.7 years of follow-up, demonstrated increased OS and PFS in patients with anaplastic oligodendroglial tumors with six cycles of adjuvant PCV chemotherapy after radiation therapy, compared with radiation therapy alone.[23][Level of evidence A1]
- OS was significantly longer in the radiation therapy and PCV arm (42.3 months vs. 30.6 months; HR, 0.75; 95% CI, 0.60–0.95).
- Patients with 1p/19q-codeleted tumors derived more benefit from adjuvant PCV chemotherapy than did those with non–1p/19q-deleted tumors.[23]
- In contrast, the RTOG trial (RTOG-9402 [NCT00002569]) demonstrated no differences in median survival by treatment arm between an 8-week, intensive PCV chemotherapy regimen followed by immediate involved-field-plus-radiation therapy and radiation therapy alone.[24]
- In an unplanned subgroup analysis, patients with 1p/19q-codeleted anaplastic oligodendrogliomas and mixed anaplastic astrocytomas demonstrated a median survival of 14.7 years versus 7.3 years (HR, 0.59; 95% CI, 0.37–0.95; P = .03).
- For patients with non-codeleted tumors, there was no difference in median survival by treatment arm (2.6 vs. 2.7 years; HR, 0.85; 95% CI, 0.58–1.23; P = .39).[24][Level of evidence A1]
- Postoperative radiation therapy alone has been compared with postoperative chemotherapy alone in patients with anaplastic gliomas (including 144 astrocytomas, 91 oligoastrocytomas, and 39 oligodendrogliomas) with crossover to the other modality at the time of tumor progression. Of the 139 patients randomly assigned to undergo radiation therapy, 135 were randomly assigned to receive chemotherapy, with a 32-week course of either PCV or single-agent temozolomide (2:1:1 randomization).[12][Levels of evidence A1 and B1]
- TTF or OS did not differ across the treatment arms and were not affected by the order of the modalities.
On the basis of these data, CODEL (NCT00887146), a study that randomly assigned patients to receive radiation therapy alone (control arm), radiation therapy with temozolomide, and temozolomide alone (exploratory arm), was halted because radiation therapy alone was no longer considered adequate treatment in patients with anaplastic oligodendroglioma with 1p/19q-codeletions.[25] Temozolomide and PCV chemotherapy in anaplastic oligodendroglioma have not been compared, although in the setting of grade III anaplastic gliomas, no survival difference was seen between PCV chemotherapy and temozolomide.[12,26]
The combination of radiation and chemotherapy is not known to be superior in outcome to sequential modality therapy.
A high proportion of anaplastic oligodendrogliomas have the IDH1 andIDH2 mutations and 1p/19q codeletion, both powerful prognostic factors for improved survival. For more information, see the Diffuse astrocytomas treatment section.[23,24] In addition, PCV chemotherapy has been shown to be predictive in a retrospective analysis of the phase III trials described earlier. Therefore, assessment of these molecular markers may aid management decisions for anaplastic oligodendrogliomas.
Mixed Gliomas Treatment
Patients with mixed glial tumors, which include oligoastrocytoma (WHO grade II) and anaplastic oligoastrocytoma (WHO grade III), have highly variable prognoses based upon their status of the IDH1 and IDH2 genes and 1p/19q chromosomes.[27,28,29] Therefore, the optimal treatment for these tumors as a group is uncertain. Often, they are treated similarly to astrocytic tumors because a subset of tumors may have outcomes similar to WHO grade III astrocytic or WHO grade IV glioblastoma tumors. Testing for these known, strong, prognostic molecular markers should be performed, which may help to guide the assessment of risk and subsequent management.
Treatment options for mixed gliomas include the following:
- Surgery plus radiation therapy with or without chemotherapy.
For more information, see the Astrocytic Tumors Treatment section.
Ependymal Tumors Treatment
Ependymal tumors (WHO grade I) and ependymomas (WHO grade II)—i.e., subependymomas and myxopapillary ependymomas—are often curable.
Treatment options for grades I and II ependymal tumors include the following:
- Surgery alone if the tumor is totally resectable.
- Surgery followed by radiation therapy to known or suspected residual tumor.
Patients with anaplastic ependymomas (WHO grade III) have variable prognoses that depend on the location and extent of disease. Frequently, but not invariably, patients with anaplastic ependymomas have worse prognoses than do those patients with lower-grade ependymal tumors.
Treatment options for anaplastic ependymomas include the following:
- Surgery plus radiation therapy.[30]
Embryonal Cell Tumors (Medulloblastomas) Treatment
Medulloblastoma occurs primarily in children but may also occur in adults.[31] For more information, see Childhood Medulloblastoma and Other Central Nervous System Embryonal Tumors Treatment.
Treatment options for medulloblastomas include the following:
- Surgery plus craniospinal radiation therapy for patients with good-risk disease.[32]
- Surgery plus craniospinal radiation therapy and various chemotherapy regimens for patients with poor-risk disease (under clinical evaluation).[32]
Pineal Parenchymal Tumors Treatment
Pineocytomas (WHO grade II), pineoblastomas (WHO grade IV), and pineal parenchymal tumors of intermediate differentiation are diverse tumors that require special consideration. Pineocytomas are slow-growing tumors and prognosis varies.
Pineoblastomas grow more rapidly and patients with these tumors have worse prognoses. Pineal parenchymal tumors of intermediate differentiation have unpredictable growth and clinical behavior.
Treatment options for pineal parenchymal tumors include the following:
- Surgery plus radiation therapy for pineocytoma.
- Surgery plus radiation therapy and chemotherapy for pineoblastoma.
Meningeal Tumors Treatment
WHO grade I meningiomas are usually curable when they are resectable. With the increasing use of sensitive neuroimaging tools, there has been more detection of asymptomatic low-grade meningiomas. Most appear to show minimal growth and can often be safely observed while therapy is deferred until growth or the development of symptoms.[33,34]
Treatment options for meningeal tumors include the following:
- Active surveillance with deferred treatment, especially for incidentally discovered asymptomatic tumors.[33,34].
- Surgery.
- Stereotactic radiosurgery for tumors smaller than 3 cm.
- Surgery plus radiation therapy in selected cases, such as for patients with known or suspected residual disease or with recurrence after previous surgery.
- Fractionated radiation therapy for patients with unresectable tumors.[35]
The prognoses for patients with WHO grade II meningiomas (atypical, clear cell, and chordoid), WHO grade III meningiomas (anaplastic/malignant, rhabdoid, and papillary), and hemangiopericytomas are worse than the prognoses for patients with low-grade meningiomas because complete resections are less commonly feasible, and the proliferative capacity is greater.
Treatment options for grades II and III meningiomas and hemangiopericytomas include the following:
- Surgery plus radiation therapy.
Germ Cell Tumors Treatment
The prognoses and treatment of patients with germ cell tumors—which include germinomas, embryonal carcinomas, choriocarcinomas, and teratomas—depend on tumor histology, tumor location, presence and amount of biological markers, and surgical resectability.
Treatment of Tumors of the Sellar Region
Craniopharyngiomas (WHO grade I) are often curable.
Treatment options for craniopharyngiomas include the following:
- Surgery alone if the tumor is totally resectable.
- Debulking surgery plus radiation therapy if the tumor is unresectable.
Treatment Options Under Clinical Evaluation for Primary CNS Tumors
Patients who have central nervous system (CNS) tumors that are either infrequently curable or unresectable should consider enrollment in clinical trials. Information about ongoing clinical trials is available from the NCI website.
Heavy-particle radiation, such as proton-beam therapy, carries the theoretical advantage of delivering high doses of ionizing radiation to the tumor bed while sparing surrounding brain tissue. The data are preliminary for this investigational technique and are not widely available.
Novel biological therapies under clinical evaluation for patients with CNS tumors include the following:[36]
- Dendritic cell vaccination.[37]
- Tyrosine kinase receptor inhibitors.[38]
- Farnesyl transferase inhibitors.
- Viral-based gene therapy.[39,40]
- Oncolytic viruses.
- Epidermal growth factor-receptor inhibitors.
- Vascular endothelial growth factor inhibitors.[36]
- Other antiangiogenesis agents.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- van den Bent MJ, Afra D, de Witte O, et al.: Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet 366 (9490): 985-90, 2005.
- Kaye AH, Walker DG: Low grade astrocytomas: controversies in management. J Clin Neurosci 7 (6): 475-83, 2000.
- Buckner JC, Pugh SL, Shaw EG, et al.: Phase III study of radiation therapy with or without procarbazine, CCNU, and vincristine (PCV) in low-grade glioma: RTOG 9802 with Alliance, ECOG, and SWOG. [Abstract] J Clin Oncol 32 (Suppl 5): A-2000, 2014.
- Buckner JC, Shaw E, Pugh S, et al.: R9802: Phase III study of radiation therapy with or without procarbazine, CCNU, and vincristine (PCV) in low-grade glioma: Results by histologic type. [Abstract] Neuro Oncol 16 (Suppl 5): A-AT-13, v11, 2014.
- Parsons DW, Jones S, Zhang X, et al.: An integrated genomic analysis of human glioblastoma multiforme. Science 321 (5897): 1807-12, 2008.
- Yan H, Parsons DW, Jin G, et al.: IDH1 and IDH2 mutations in gliomas. N Engl J Med 360 (8): 765-73, 2009.
- Dubbink HJ, Taal W, van Marion R, et al.: IDH1 mutations in low-grade astrocytomas predict survival but not response to temozolomide. Neurology 73 (21): 1792-5, 2009.
- Sanson M, Marie Y, Paris S, et al.: Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27 (25): 4150-4, 2009.
- Hartmann C, Hentschel B, Wick W, et al.: Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol 120 (6): 707-18, 2010.
- Hartmann C, Meyer J, Balss J, et al.: Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118 (4): 469-74, 2009.
- Watanabe T, Nobusawa S, Kleihues P, et al.: IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174 (4): 1149-53, 2009.
- Wick W, Hartmann C, Engel C, et al.: NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol 27 (35): 5874-80, 2009.
- Hegi ME, Diserens AC, Gorlia T, et al.: MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352 (10): 997-1003, 2005.
- Gilbert MR, Wang M, Aldape KD, et al.: Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol 31 (32): 4085-91, 2013.
- Stupp R, Mason WP, van den Bent MJ, et al.: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352 (10): 987-96, 2005.
- Stupp R, Hegi ME, Mason WP, et al.: Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10 (5): 459-66, 2009.
- Gilbert MR, Dignam JJ, Armstrong TS, et al.: A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370 (8): 699-708, 2014.
- Chinot OL, Wick W, Mason W, et al.: Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 370 (8): 709-22, 2014.
- Fallon KB, Palmer CA, Roth KA, et al.: Prognostic value of 1p, 19q, 9p, 10q, and EGFR-FISH analyses in recurrent oligodendrogliomas. J Neuropathol Exp Neurol 63 (4): 314-22, 2004.
- Smith JS, Perry A, Borell TJ, et al.: Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol 18 (3): 636-45, 2000.
- Okamoto Y, Di Patre PL, Burkhard C, et al.: Population-based study on incidence, survival rates, and genetic alterations of low-grade diffuse astrocytomas and oligodendrogliomas. Acta Neuropathol 108 (1): 49-56, 2004.
- van den Bent MJ, Chinot O, Boogerd W, et al.: Second-line chemotherapy with temozolomide in recurrent oligodendroglioma after PCV (procarbazine, lomustine and vincristine) chemotherapy: EORTC Brain Tumor Group phase II study 26972. Ann Oncol 14 (4): 599-602, 2003.
- van den Bent MJ, Brandes AA, Taphoorn MJ, et al.: Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 31 (3): 344-50, 2013.
- Cairncross G, Wang M, Shaw E, et al.: Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol 31 (3): 337-43, 2013.
- Gilbert MR: Minding the Ps and Qs: perseverance and quality studies lead to major advances in patients with anaplastic oligodendroglioma. J Clin Oncol 31 (3): 299-300, 2013.
- Brada M, Stenning S, Gabe R, et al.: Temozolomide versus procarbazine, lomustine, and vincristine in recurrent high-grade glioma. J Clin Oncol 28 (30): 4601-8, 2010.
- Jiao Y, Killela PJ, Reitman ZJ, et al.: Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 3 (7): 709-22, 2012.
- Killela PJ, Reitman ZJ, Jiao Y, et al.: TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A 110 (15): 6021-6, 2013.
- Killela PJ, Pirozzi CJ, Healy P, et al.: Mutations in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget 5 (6): 1515-25, 2014.
- Oya N, Shibamoto Y, Nagata Y, et al.: Postoperative radiotherapy for intracranial ependymoma: analysis of prognostic factors and patterns of failure. J Neurooncol 56 (1): 87-94, 2002.
- Brandes AA, Ermani M, Amista P, et al.: The treatment of adults with medulloblastoma: a prospective study. Int J Radiat Oncol Biol Phys 57 (3): 755-61, 2003.
- Brandes AA, Franceschi E, Tosoni A, et al.: Long-term results of a prospective study on the treatment of medulloblastoma in adults. Cancer 110 (9): 2035-41, 2007.
- Nakamura M, Roser F, Michel J, et al.: The natural history of incidental meningiomas. Neurosurgery 53 (1): 62-70; discussion 70-1, 2003.
- Yano S, Kuratsu J; Kumamoto Brain Tumor Research Group: Indications for surgery in patients with asymptomatic meningiomas based on an extensive experience. J Neurosurg 105 (4): 538-43, 2006.
- Debus J, Wuendrich M, Pirzkall A, et al.: High efficacy of fractionated stereotactic radiotherapy of large base-of-skull meningiomas: long-term results. J Clin Oncol 19 (15): 3547-53, 2001.
- Fine HA: Promising new therapies for malignant gliomas. Cancer J 13 (6): 349-54, 2007 Nov-Dec.
- Fecci PE, Mitchell DA, Archer GE, et al.: The history, evolution, and clinical use of dendritic cell-based immunization strategies in the therapy of brain tumors. J Neurooncol 64 (1-2): 161-76, 2003 Aug-Sep.
- Newton HB: Molecular neuro-oncology and development of targeted therapeutic strategies for brain tumors. Part 1: Growth factor and Ras signaling pathways. Expert Rev Anticancer Ther 3 (5): 595-614, 2003.
- Kew Y, Levin VA: Advances in gene therapy and immunotherapy for brain tumors. Curr Opin Neurol 16 (6): 665-70, 2003.
- Chiocca EA, Aghi M, Fulci G: Viral therapy for glioblastoma. Cancer J 9 (3): 167-79, 2003 May-Jun.
Treatment of Primary Tumors of the Spinal Axis
Surgery and radiation therapy are the primary modalities used to treat tumors of the spinal axis; therapeutic options vary according to the histology of the tumor.[1] The experience with chemotherapy for primary spinal cord tumors is limited; no reports of controlled clinical trials are available for these types of tumors.[1,2] Chemotherapy is indicated for most patients with leptomeningeal involvement from a primary or metastatic tumor and positive cerebrospinal fluid cytology.[1] Most patients require treatment with corticosteroids, particularly if they are receiving radiation therapy.
Patients who have spinal axis tumors that are either infrequently curable or unresectable should consider enrollment in clinical trials. Information about ongoing clinical trials is available from the NCI website.
References:
- Cloughesy T, Selch MT, Liau L: Brain. In: Haskell CM: Cancer Treatment. 5th ed. WB Saunders Co, 2001, pp 1106-42.
- Mehta M, Vogelbaum MA, Chang S, et al.: Neoplasms of the central nervous system. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Lippincott Williams & Wilkins, 2011, pp 1700-49.
Metastatic Brain Tumors
General Information About Metastatic Brain Tumors
Brain metastases outnumber primary neoplasms by at least 10 to 1, and they occur in 20% to 40% of cancer patients, with subsequent median survival generally less than 6 months.[1] The exact incidence is unknown because no national cancer registry documents brain metastases, but it has been estimated that 98,000 to 170,000 new cases are diagnosed in the United States each year.[2,3] This number may be increasing because of the capacity of magnetic resonance imaging (MRI) to detect small metastases and because of prolonged survival resulting from improved systemic therapy.[1,2]
The most common primary tumors with brain metastases and the percentage of patients affected are as follows:[1,2]
- Lung (18%–64%).
- Breast (2%–21%).
- Cancer of unknown primary (1%–18%).
- Melanoma (4%–16%).
- Colorectal (2%–12%).
- Kidney (1%–8%).
Eighty percent of brain metastases occur in the cerebral hemispheres, 15% occur in the cerebellum, and 5% occur in the brain stem.[2] Metastases to the brain are multiple in more than 70% of cases, but solitary metastases also occur.[1]
Brain involvement can occur with cancers of the nasopharyngeal region by direct extension along the cranial nerves or through the foramina at the base of the skull. Dural metastases may constitute as much as 9% of total brain metastases.
Clinical Features
The diagnosis of brain metastases in cancer patients is based on the following:
- Patient history.
- Neurological examination.
- Diagnostic procedures, including a contrast MRI of the brain.
Patients may describe any of the following:
- Headaches.
- Weakness.
- Seizures.
- Sensory defects.
- Gait problems.
Often, family members or friends may notice the following:
- Lethargy.
- Emotional lability.
- Personality change.
Diagnostic Evaluation
A physical examination may show objective neurological findings or only minor cognitive changes. The presence of multiple lesions and a high predilection of primary tumor metastasis may be sufficient to make the diagnosis of brain metastasis.
A lesion in the brain should not be assumed to be a metastasis just because a patient has had a previous cancer; such an assumption could result in overlooking appropriate treatment of a curable tumor.
Imaging tests
Computed tomography scans with contrast or MRIs with gadolinium are quite sensitive in diagnosing the presence of metastases. Positron emission tomography scanning and spectroscopic evaluation are new strategies to diagnose cerebral metastases and to differentiate the metastases from other intracranial lesions.[4]
Biopsy
In the case of a solitary lesion or a questionable relationship to the primary tumor, a brain biopsy (via resection or stereotactic biopsy) may be necessary.
Treatment of Metastatic Brain Tumors
The optimal therapy for patients with brain metastases continues to evolve.[1,2,5] The following treatments have been used in the management of metastatic brain tumors:
- Radiation therapy.
- Radiosurgery.
- Surgical resection.
- Corticosteroids.
- Anticonvulsants.
Because most cases of brain metastases involve multiple metastases, a mainstay of therapy has historically been whole-brain radiation therapy (WBRT). However, stereotactic radiosurgery has become increasingly common. The role of radiosurgery continues to be defined. Stereotactic radiosurgery in combination with WBRT has been assessed.
Surgery is indicated to obtain tissue from a metastasis with an unknown primary tumor or to decompress a symptomatic dominant lesion that is causing significant mass effect.
Chemotherapy is usually not the primary therapy for most patients; however, it may have a role in the treatment of patients with brain metastases from chemosensitive tumors and can even be curative when combined with radiation for metastatic testicular germ cell tumors.[1,6] Intrathecal chemotherapy is also used for meningeal spread of metastatic tumors.
Treatment for patients with one to four metastases
Treatment options for patients with one to four metastases
About 10% to 15% of patients with cancer will have a single brain metastasis. Radiation therapy is the mainstay of palliation for these patients. The extent of extracranial disease can influence treatment of the brain lesions. In the presence of extensive active systemic disease, surgery provides little benefit for overall survival (OS). In patients with stable minimal extracranial disease, combined modality treatment may be considered, using surgical resection followed by radiation therapy. However, the published literature does not provide clear guidance.
Treatment options for patients with one to four metastases include the following:
- WBRT with or without surgical resection.
- WBRT with or without stereotactic radiosurgery.
- Focal therapy alone (surgical resection or stereotactic radiosurgery).
Evidence (treatment for one to four metastases):
- Three randomized trials examined resection of solitary brain metastases followed by WBRT versus WBRT alone, totaling 195 randomly assigned patients.[7,8,9] The process that necessarily goes into selecting appropriate patients for surgical resection may account for the small numbers in each trial. In the first trial,[7][Level of evidence B1] performed at a single center, all patients were selected and operated upon by one surgeon.
- The first two trials showed an improvement in survival in the surgery group,[7,8] but the third trial showed a trend in favor of the WBRT-only group.[9]
- The three trials were combined in a trial-level meta-analysis.[10] The combined analysis showed the following:
- The combined analysis did not show a statistically significant difference in OS (hazard ratio [HR], 0.72; 95% confidence interval [CI], 0.34–1.53; P = .4); or in death from neurological causes (relative riskdeath, 0.68; 95% CI, 0.43–1.09; P = .11).[10]
- One of the trials reported that combined therapy increased the duration of functionally independent survival.[7][Level of evidence B1]
- None of the trials assessed or reported quality of life.
- The need for WBRT after resection of solitary brain metastases has been studied.[11] Patients were randomly assigned to either undergo postoperative WBRT or receive no further treatment after resection.
- Patients in the WBRT group were less likely to have tumor progression in the brain and were significantly less likely to die of neurological causes.
- OS was the same in each group, and there was no difference in duration of functional independence.
- One additional randomized study of observation versus WBRT after either surgery or stereotactic radiosurgery for solitary brain metastases was closed after 19 patients had been entered because of slow accrual; therefore, little can be deduced from the trial.[12]
- A Radiation Therapy Oncology Group (RTOG) study (RTOG-9508) randomly assigned 333 patients with one to three metastases with a maximum diameter of 4 cm to WBRT (37.5 Gy over 3 weeks) with or without a stereotactic boost.[13] Patients with active systemic disease requiring therapy were excluded. The primary end point was OS with predefined hypotheses in both the full study population and the 186 patients with a solitary metastasis (and no statistical adjustment of P values for the two separate hypotheses).[13][Levels of evidence B1 for the full study population and A1 for patients with solitary metastases]
- Mean OS in the combined-therapy group was 5.7 months, and mean OS in the WBRT–alone group was 6.5 months (P = .14).
- In the subgroup with solitary metastases, OS was better in the combined-therapy group (6.5 months vs. 4.9 months; P = .039 in univariate analysis; P = .053 in a multivariable analysis adjusting for baseline prognostic factors).
- In patients with multiple metastases, survival was 5.8 months in the combined-therapy group versus 6.7 months in the WBRT–only group (P = .98).
- The combined-treatment group had a survival advantage of 2.5 months in patients with a single metastasis but not in patients with multiple lesions.
- Local control was better in the full population with combined therapy.
- At the 6-month follow-up, Karnofsky Performance status (considered a soft end point because of its imprecision and subjectivity) was better in the combined-therapy group, but there was no difference in mental status between the treatment groups. Acute and late toxicities were similar in both treatment arms. Quality of life was not assessed.
- Mean OS in the combined-therapy group was 5.7 months, and mean OS in the WBRT–alone group was 6.5 months (P = .14).
- A phase III randomized trial compared adjuvant WBRT with observation after surgery or radiosurgery for a limited number of brain metastases in patients with stable solid tumors.[14][Level of evidence A3]
- Health-related quality of life was improved in the observation-only arm, compared with WBRT.
- Patients in the observation arm had better mean scores in physical, role, and cognitive functioning at 9 months.
- In an exploratory analysis, statistically significant worse scores for bladder control, communication deficit, drowsiness, hair loss, motor dysfunction, leg weakness, appetite loss, constipation, nausea/vomiting, pain, and social functioning were observed in patients who underwent WBRT, compared with those who underwent observation only.
- A meta-analysis of two trials with a total of 358 participants found no statistically significant difference in OS between the WBRT plus stereotactic radiosurgery group and the WBRT-alone group (HR, 0.82; 95% CI, 0.65–1.02).[15][Level of evidence B1]
- Patients in the WBRT plus stereotactic radiosurgery group had decreased local failure, compared with patients who received WBRT alone (HR, 0.27; 95% CI, 0.14–0.52).
- Unchanged or improved Karnofsky Performance status at 6 months was seen in 43% of patients in the combined-therapy group versus 28% in the WBRT-alone group (P = .03).
A study that had a primary end point of learning and neurocognition, using a standardized test for total recall, was stopped by the Data and Safety Monitoring Board because of worse outcomes in the WBRT group.[16][Level of evidence B1]
Given this body of information, focal therapy plus WBRT or focal therapy alone, with close follow-up with serial MRIs and initiation of salvage therapy when clinically indicated, appear to be reasonable treatment options. The pros and cons of each approach should be discussed with the patient.
Several randomized trials have been performed that were designed with varying primary end points to address whether WBRT is necessary after focal treatment. The results can be summarized as follows:[16,17,18]
- Studies consistently show that the addition of WBRT to focal therapy decreases the risk of progression and new metastases in the brain.
- The addition of WBRT does not improve OS.
- The decrease in risk of intracranial disease progression does not translate into improved functional or neurological status, nor does it appear to decrease the risk of death from neurological deterioration.
- About one-half or more of the patients who receive focal therapy alone ultimately require salvage therapy, such as WBRT or radiosurgery, compared with about one-quarter of the patients who are given up-front WBRT.
- The impact of better local control associated with WBRT on quality of life has not been reported and remains an open question.
Leptomeningeal Carcinomatosis (LC)
LC occurs in about 5% of all cancer patients. The most common types of cancer to spread to the leptomeninges are:
- Breast tumors (35%).
- Lung tumors (24%).
- Hematologic malignancies (16%).
Diagnosis includes a combination of neurospinal axis imaging and cerebrospinal fluid (CSF) cytology. Median OS is in the range of 10 to 12 weeks.
The management of LC includes the following:
- Intrathecal chemotherapy.
- Intrathecal chemotherapy and systemic chemotherapy.
- Intrathecal chemotherapy and radiation therapy.
- Supportive care.
In a series of 149 patients with metastatic non-small cell lung carcinoma, cytologically proven LC, poor performance status, high protein level in the CSF, and a high initial CSF white blood cell count were significant poor prognostic factors for survival.[19] Patients received active treatment, including intrathecal chemotherapy, WBRT, or epidermal growth factor receptor-tyrosine kinase inhibitors, or underwent a ventriculoperitoneal shunt procedure.
In a retrospective series of 38 patients with metastatic breast cancer and LC, the proportion of LC cases varied by breast cancer subtype:[20]
- Luminal A (18.4%).
- Luminal B (31.6%).
- Human epidermal growth factor receptor 2 (HER2)-positive (26.3%).
- Triple-negative breast cancer subtype (23.7%).
Patients with triple-negative breast cancer had a shorter interval between metastatic breast cancer diagnosis and the development of LC. Median survival did not differ across breast cancer subtypes. Consideration of intrathecal administration of trastuzumab in patients with HER2-positive LC has also been described in case reports.[21]
References:
- Patchell RA: The management of brain metastases. Cancer Treat Rev 29 (6): 533-40, 2003.
- Mehta M, Vogelbaum MA, Chang S, et al.: Neoplasms of the central nervous system. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Lippincott Williams & Wilkins, 2011, pp 1700-49.
- Hutter A, Schwetye KE, Bierhals AJ, et al.: Brain neoplasms: epidemiology, diagnosis, and prospects for cost-effective imaging. Neuroimaging Clin N Am 13 (2): 237-50, x-xi, 2003.
- Schaefer PW, Budzik RF, Gonzalez RG: Imaging of cerebral metastases. Neurosurg Clin N Am 7 (3): 393-423, 1996.
- Soffietti R, Cornu P, Delattre JY, et al.: EFNS Guidelines on diagnosis and treatment of brain metastases: report of an EFNS Task Force. Eur J Neurol 13 (7): 674-81, 2006.
- Ogawa K, Yoshii Y, Nishimaki T, et al.: Treatment and prognosis of brain metastases from breast cancer. J Neurooncol 86 (2): 231-8, 2008.
- Patchell RA, Tibbs PA, Walsh JW, et al.: A randomized trial of surgery in the treatment of single metastases to the brain. N Engl J Med 322 (8): 494-500, 1990.
- Vecht CJ, Haaxma-Reiche H, Noordijk EM, et al.: Treatment of single brain metastasis: radiotherapy alone or combined with neurosurgery? Ann Neurol 33 (6): 583-90, 1993.
- Mintz AH, Kestle J, Rathbone MP, et al.: A randomized trial to assess the efficacy of surgery in addition to radiotherapy in patients with a single cerebral metastasis. Cancer 78 (7): 1470-6, 1996.
- Hart MG, Grant R, Garside R, et al.: Chemotherapeutic wafers for high grade glioma. Cochrane Database Syst Rev (3): CD007294, 2008.
- Patchell RA, Tibbs PA, Regine WF, et al.: Postoperative radiotherapy in the treatment of single metastases to the brain: a randomized trial. JAMA 280 (17): 1485-9, 1998.
- Roos DE, Wirth A, Burmeister BH, et al.: Whole brain irradiation following surgery or radiosurgery for solitary brain metastases: mature results of a prematurely closed randomized Trans-Tasman Radiation Oncology Group trial (TROG 98.05). Radiother Oncol 80 (3): 318-22, 2006.
- Andrews DW, Scott CB, Sperduto PW, et al.: Whole brain radiation therapy with or without stereotactic radiosurgery boost for patients with one to three brain metastases: phase III results of the RTOG 9508 randomised trial. Lancet 363 (9422): 1665-72, 2004.
- Soffietti R, Kocher M, Abacioglu UM, et al.: A European Organisation for Research and Treatment of Cancer phase III trial of adjuvant whole-brain radiotherapy versus observation in patients with one to three brain metastases from solid tumors after surgical resection or radiosurgery: quality-of-life results. J Clin Oncol 31 (1): 65-72, 2013.
- Patil CG, Pricola K, Sarmiento JM, et al.: Whole brain radiation therapy (WBRT) alone versus WBRT and radiosurgery for the treatment of brain metastases. Cochrane Database Syst Rev 9: CD006121, 2012.
- Chang EL, Wefel JS, Hess KR, et al.: Neurocognition in patients with brain metastases treated with radiosurgery or radiosurgery plus whole-brain irradiation: a randomised controlled trial. Lancet Oncol 10 (11): 1037-44, 2009.
- Aoyama H, Shirato H, Tago M, et al.: Stereotactic radiosurgery plus whole-brain radiation therapy vs stereotactic radiosurgery alone for treatment of brain metastases: a randomized controlled trial. JAMA 295 (21): 2483-91, 2006.
- Kocher M, Soffietti R, Abacioglu U, et al.: Adjuvant whole-brain radiotherapy versus observation after radiosurgery or surgical resection of one to three cerebral metastases: results of the EORTC 22952-26001 study. J Clin Oncol 29 (2): 134-41, 2011.
- Lee SJ, Lee JI, Nam DH, et al.: Leptomeningeal carcinomatosis in non-small-cell lung cancer patients: impact on survival and correlated prognostic factors. J Thorac Oncol 8 (2): 185-91, 2013.
- Torrejón D, Oliveira M, Cortes J, et al.: Implication of breast cancer phenotype for patients with leptomeningeal carcinomatosis. Breast 22 (1): 19-23, 2013.
- Bartsch R, Berghoff AS, Preusser M: Optimal management of brain metastases from breast cancer. Issues and considerations. CNS Drugs 27 (2): 121-34, 2013.
Treatment of Recurrent Adult CNS Tumors
Patients who have recurrent CNS tumors are rarely curable and should consider enrollment in clinical trials. Information about ongoing clinical trials is available from the NCI website.
Treatment options for recurrent CNS tumors include the following:
- Chemotherapy.
- Antiangiogenesis therapy.
- Radiation therapy.
- Surgery.
Chemotherapy
Localized chemotherapy (carmustine wafer)
Carmustine wafers have been investigated for the treatment of recurrent malignant gliomas, but the impact on survival is less clear than at the time of initial diagnosis and resection.
Evidence (localized chemotherapy):
- In a multicenter, randomized, placebo-controlled trial, 222 patients with recurrent malignant primary brain tumors requiring reoperation were randomly assigned to receive implanted carmustine wafers or placebo biodegradable wafers.[1][Level of evidence A1] Approximately one-half of the patients had received previous systemic chemotherapy. The two treatment groups were well balanced at baseline.
- Median survival was 31 weeks in the group receiving carmustine wafers versus 23 weeks in the group receiving placebo wafers. The statistical significance between the two overall survival curves depended on the method of analysis.
- The hazard ratio (HR) for risk of dying in the direct intention-to-treat comparison between the two groups was 0.83 (95% confidence interval [CI], 0.63–1.10; P = .19). The baseline characteristics were similar in the two groups, but the investigators performed an additional analysis, adjusting for prognostic factors, because they felt that even small differences in baseline characteristics could have a powerful influence on outcomes. In the adjusted proportional hazards model, the HR for risk of death was 0.67 (95% CI, 0.51–0.90,; P = .006). The investigators emphasized this latter analysis and reported this as a positive trial.[1][Level of evidence A1]
- A Cochrane Collaboration systematic review of chemotherapeutic wafers for high-grade glioma focused on the unadjusted analysis and reported the same trial as negative.[2]
Systemic chemotherapy
Systemic therapy (e.g., temozolomide, lomustine, or the combination of procarbazine, a nitrosourea, and vincristine (PCV) in patients who have not previously received the drugs) has been used at the time of recurrence of primary malignant brain tumors. However, this approach has not been tested in controlled studies. Patient-selection factors likely play a strong role in determining outcomes, so the impact of therapy on survival is not clear.
Antiangiogenesis Therapy
In 2009, the U.S. Food and Drug Administration (FDA) granted accelerated approval of bevacizumab monotherapy for patients with progressive glioblastoma. The indication was granted under the FDA's accelerated approval program that permits the use of certain surrogate end points or an effect on a clinical end point other than survival or irreversible morbidity as bases for approvals of products intended for serious or life-threatening illnesses or conditions.
The approval was based on the demonstration of improved objective response rates observed in two historically controlled, single-arm, or noncomparative phase II trials.[3,4][Level of evidence C3] On the basis of these data and FDA approval, bevacizumab monotherapy has become standard therapy for recurrent glioblastoma.
Evidence (antiangiogenesis therapy):
- The FDA independently reviewed an open-label, multicenter, noncomparative phase II study that randomly assigned 167 patients with recurrent glioblastoma multiforme (GBM) to receive bevacizumab alone or bevacizumab in combination with irinotecan,[3] although only efficacy data from the bevacizumab monotherapy arm (n = 85) were used to support drug approval.
- Tumor responses were observed in 26% of patients treated with bevacizumab alone, and the median duration of response in these patients was 4.2 months.
- On the basis of this externally controlled trial, the incidence of adverse events associated with bevacizumab did not appear to be significantly increased in GBM patients.
- The FDA independently assessed another single-arm, single-institution trial in which 56 patients with recurrent glioblastoma were treated with bevacizumab alone.[4]
- Responses were observed in 20% of patients, and the median duration of response was 3.9 months.
No data are available from prospective randomized controlled trials demonstrating improvement in health outcomes, such as disease-related symptoms or increased survival with the use of bevacizumab to treat glioblastoma.
Radiation Therapy
Because there are no randomized trials, the role of repeat radiation after disease progression or the development of radiation-induced cancers is also ill defined. Interpretation is difficult because the literature is limited to small retrospective case series.[5] The decision must be made carefully because of the risk of neurocognitive deficits and radiation necrosis.
Surgery
Re-resection of recurrent CNS tumors is an option for some patients. However, most patients do not qualify because of a deteriorating condition or technically inoperable tumors. The evidence is limited to noncontrolled studies and case series of patients who are healthy enough and have tumors that are small enough to technically debulk. The impact on survival of reoperation versus patient selection is not known.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Brem H, Piantadosi S, Burger PC, et al.: Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-brain Tumor Treatment Group. Lancet 345 (8956): 1008-12, 1995.
- Hart MG, Grant R, Garside R, et al.: Chemotherapeutic wafers for high grade glioma. Cochrane Database Syst Rev (3): CD007294, 2008.
- Friedman HS, Prados MD, Wen PY, et al.: Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27 (28): 4733-40, 2009.
- Kreisl TN, Kim L, Moore K, et al.: Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol 27 (5): 740-5, 2009.
- Paulino AC, Mai WY, Chintagumpala M, et al.: Radiation-induced malignant gliomas: is there a role for reirradiation? Int J Radiat Oncol Biol Phys 71 (5): 1381-7, 2008.
Latest Updates to This Summary (03 / 06 / 2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
General Information About Adult Central Nervous System Tumors
Updated statistics with estimated new cases and deaths for 2024 (cited American Cancer Society as reference 2).
Updated text about incidence and mortality rates.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of adult central nervous system tumors. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewer for Adult Central Nervous System Tumors Treatment is:
- Minh Tam Truong, MD (Boston University Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Adult Central Nervous System Tumors Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/brain/hp/adult-brain-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389419]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2024-03-06


This information does not replace the advice of a doctor. Ignite Healthwise, LLC disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the Terms of Use and Privacy Policy. Learn how we develop our content.
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.